Michelson Diagnostics
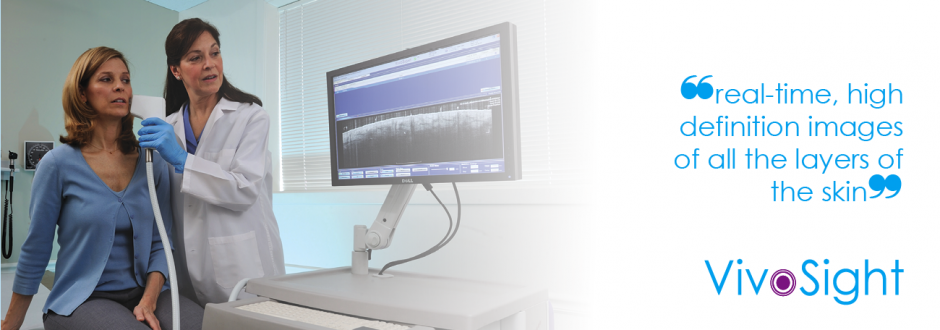
Developer of the VivoSight® OCT scanner



Newsletter

Subscribe to our Newsletter
Sector

We have developed a world-leading patented medical imaging technology
Product

For more information on our VivoSight OCT System, click here to visit www.vivosight.com